- Article

Rising Burden of Potentially Inherited Arrhythmic Syndromes and Sudden Cardiac Death in the United States, 1999–2024
- Faizan Ahmed,
- Swapnil Patel and
- Mohammad Amir Hossain
- + 9 authors
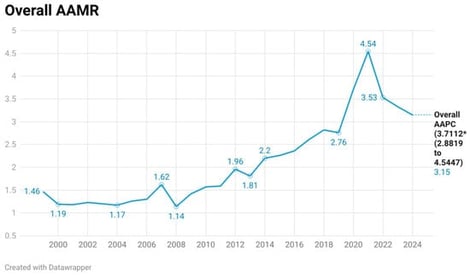
Background: Inherited arrhythmic syndromes (IAS) are an important but under-recognized cause of sudden cardiac death (SCD), particularly in younger individuals. Understanding long-term mortality trends is essential to evaluate their public health impact. Objective: To assess temporal trends and demographic disparities of IAS-related sudden cardiac death among individuals aged 5–44 years in the United States using CDC WONDER data. Methods: This retrospective observational study utilized the CDC WONDER Multiple Cause-of-Death database from 1999 to 2024. Deaths were identified using ICD-10 codes for non-ischemic arrhythmogenic conditions (I42, I44, I45, I47, I49) in combination with sudden cardiac arrest or unexplained death (I46, R96). Ischemic heart disease (I20–I25) was excluded to enhance specificity for inherited causes. Crude and age-adjusted mortality rates (AAMRs) per 1,000,000 population were calculated and stratified by age, sex, race/ethnicity, region, urbanization, and place of death. Joinpoint software helped us calculate the average annual percentage change (AAPC)/annual percent change (APC) in AAMRs and the 95% CIs for these changes. Results: A total of 8879 deaths were identified over the study period. The AAMR increased from 1.46 (95% CI: 1.27–1.64) in 1999 to 3.15 (95% CI: 2.89–3.42) in 2024, peaking at 4.54 in 2021, with an overall AAPC of 3.71% (p < 0.000001). Mortality was higher in males; however, females demonstrated a greater relative increase over time. Non-Hispanic Black individuals exhibited the highest mortality rates and fastest rise. The 25–44-year age group accounted for most deaths and showed the steepest increase. Regional and urban–rural disparities were observed, with higher mortality rates in the South and rural areas. Conclusions: It is concluded that mortality related to inherited arrhythmic syndromes and sudden cardiac death is rising among young individuals in the United States. The findings highlight a growing burden of potentially inherited arrhythmogenic conditions and underscore the need for early detection strategies, including genetic screening and targeted public health interventions, to reduce premature cardiovascular mortality.
2 July 2026